




L’hémophilie
Un peu d’Histoire …

Evolution de l’espérance de vie d’un hémophile :
- En 1900 – 11ans
- En 1950 – 25ans
- En 1980 – 63 ans
- Aujourd’hui – tend à la normale
Définition+
L’Hémophilie est une maladie hémorragique génétique due à l’absence totale ou partielle d’un facteur de coagulation :
- S’il s’agit du facteur VIII (8) on parle d’hémophilie A
- S’il s’agit du facteur F IX (9) on parle d’hémophilie B
La prévalence est d’environ 1/7500 naissances de sexe masculin pour l’hémophilie A et d’1/30000 pour l’hémophilie B. L’hémophilie A représente environ 80% des hémophiles.
On compte en France environ 7500 hémophiles ; c’est la plus fréquente des maladies hémorragiques graves.
L’expression de l’hémophilie est variable selon le taux de facteur circulant, on distingue plusieurs formes :
- Hémophilie sévère : taux de FVIII ou de FIX < 1%
- Hémophilie modérée : taux de FVIII ou de FIX entre 1 et 5%
- Hémophilie mineure : taux de FVIII ou de FIX entre 5 et 40%
Transmission de l’hémophilie
Le FVIII et le FIX sont des protéines de la coagulation. Chacune est synthétisée par 1 gène situé sur le chromosome X. Ce déficit est lié à des mutations génétiques de ce chromosome X. La transmission de l’hémophilie est récessive et liée à l’X.
Cette maladie touche essentiellement les garçons alors que les femmes porteuses d’une mutation sur l’un de leurs 2 chromosomes X sont dites conductrices et la plupart du temps leur taux de facteur de coagulation est suffisant pour leur éviter des manifestations hémorragiques. Toutefois, certaines femmes présentent un taux bas en raison d’évènements génétiques particuliers et leurs signes hémorragiques peuvent être significatifs : elles sont alors considérées comme hémophiles dont le niveau de sévérité est variable.

Transmission de le cas d’une femme conductrice
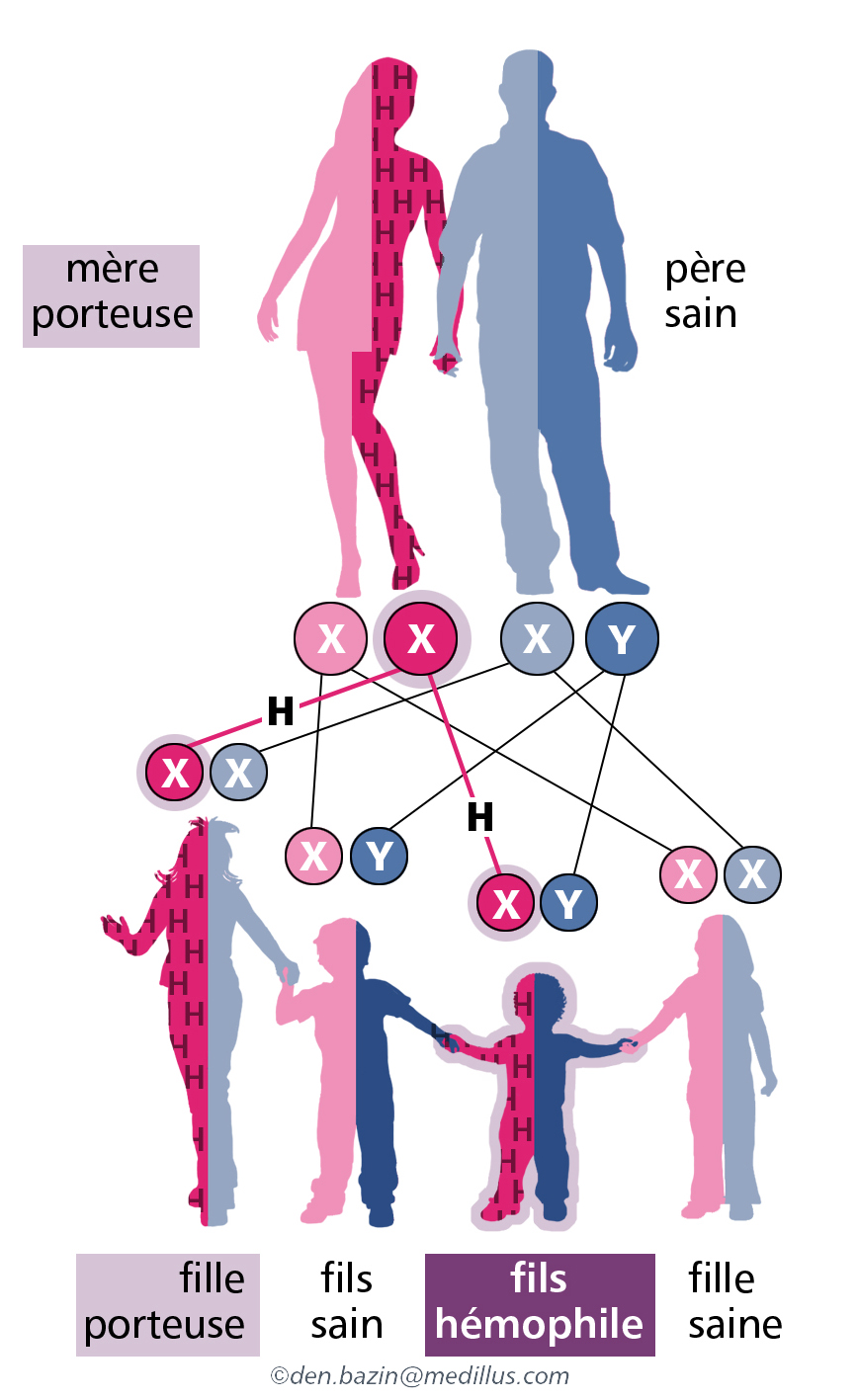
1 fille sur 2 sera conductrice et 1 garçon sur 2 sera hémophile
Transmission dans le cas d’une homme atteint d’hémophilie

Toutes les filles seront porteuses de la maladie= conductrices
Dans 30% des cas, il n’y a pas d’antécédents familiaux d’hémophilie : on parle alors d’une néo-mutation ou mutation de novo. Cette mutation nouvellement apparue peut avoir eu lieu dans l’ovule de la mère ou dans le spermatozoïde du père, ou plus tard chez le fœtus lui-même. Cette mutation sera transmissible à la descendance.
Coagulation et hémophilie
Lors d’une brèche vasculaire ou d’un traumatisme, un saignement plus ou moins important se produit. Le processus de coagulation s’active : les plaquettes, le facteur Willebrand et les différents facteurs de coagulation s’activent les uns les autres, en cascade pour former un caillot solide et arrêter le saignement.
Pour plus d’informations sur l’hémostase, cliquer ici.
Dans le cas de l’hémophilie, un de ces facteurs : le VIII ou le IX est absent ou en quantité insuffisante pour permettre la formation d’un caillot solide et l’arrêt du saignement.
Chez les hémophiles, les saignements ne sont pas plus importants mais ils durent plus longtemps.
Manifestations de l’hémophilie+
S’il n’y a pas d’antécédents familiaux, la découverte de la maladie se fera au cours de la 1ère année de l’enfant (pour la forme sévère), lorsqu’il va commencer à se déplacer avec l’apparition de saignements superficiels : ecchymoses, hématomes…
Pour les formes modérées et mineurs, les symptômes pourront être plus tardifs ou diagnostiqués avant une intervention chirurgicale.
Les manifestations de l’hémophilie sont des accidents hémorragiques plus ou moins importants au niveau du corps.
Selon la sévérité de la maladie les manifestations seront différentes :
- Hémophilie sévère :
- saignements « spontanés » (sans cause identifiée) au niveau des articulations (hémarthroses) ou des muscles (hématomes)
- saignements traumatiques, liés à un choc ou une chirurgie
- Hémophilie modérée : les saignements sont moins spontanés et le plus souvent traumatiques, liés à une chirurgie
- Hémophilie mineure : les saignements sont traumatiques ou liés à une intervention chirurgicale
Symptômes de l’hémophilie :
- Ecchymoses ou « bleus » : présence de sang en petite quantité sous la peau, fréquents chez les enfants mais souvent sans gravité.
- Hématomes musculaires : saignements au niveau des muscles. Ils peuvent apparaitre sur tout le corps, forment une « boule/bosse » et son généralement douloureux. Selon leur localisation, les hématomes peuvent être plus ou moins dangereux en raison du risque de compression vasculo-nerveuse (au niveau de la main, de l’avant-bras, du quadriceps et du mollet…).
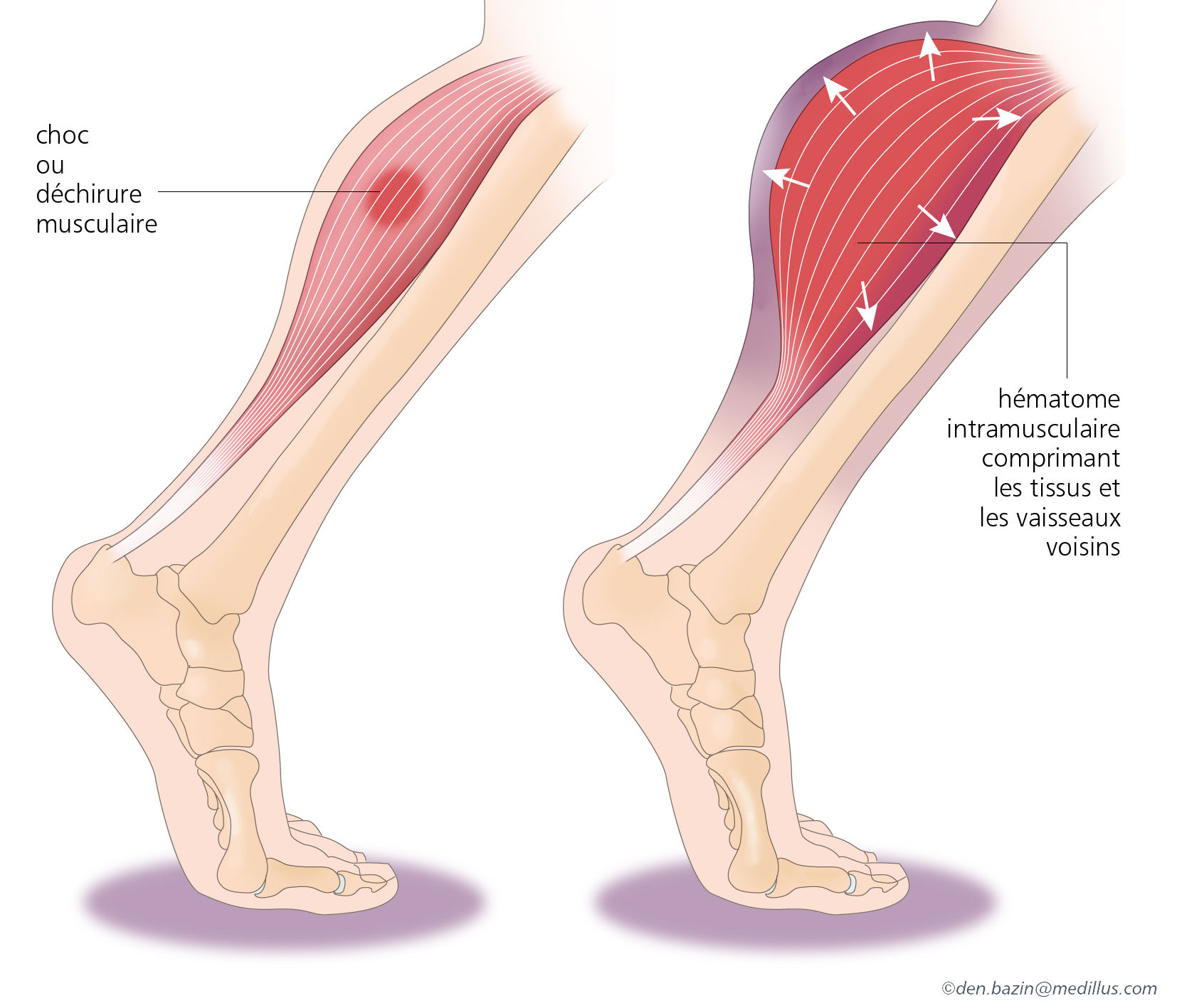
Formation d’un hématome (cliquer pour agrandir)

Les localisations dangereuses (cliquer pour agrandir)
- Hémarthroses : saignements au niveau des articulations. Elles peuvent être spontanées (dans les formes sévères de la maladie et parfois les formes modérés) ou traumatiques (pour les formes modérées et mineures). Toutes les articulations peuvent être touchées mais certaines sont plus fréquentes comme les genoux, les chevilles, les coudes ; d’autres comme l’hémarthrose de hanche sont moins fréquentes mais plus à risque de complications. Les signes d’une hémarthrose sont une sensation de compression, un gonflement de l’articulation, une douleur et une gêne à la mobilité de cette articulation. La précocité du traitement permettra une résorption plus rapide de l’hémarthrose et de limiter les complications qui peuvent en découler. Les hémarthroses peuvent survenir à plusieurs reprises sur une même articulation (= articulation cible) (définir articulation cible) et peuvent évoluer vers une arthropathie.

Formation d’une hémarthrose (cliquer pour agrandir)
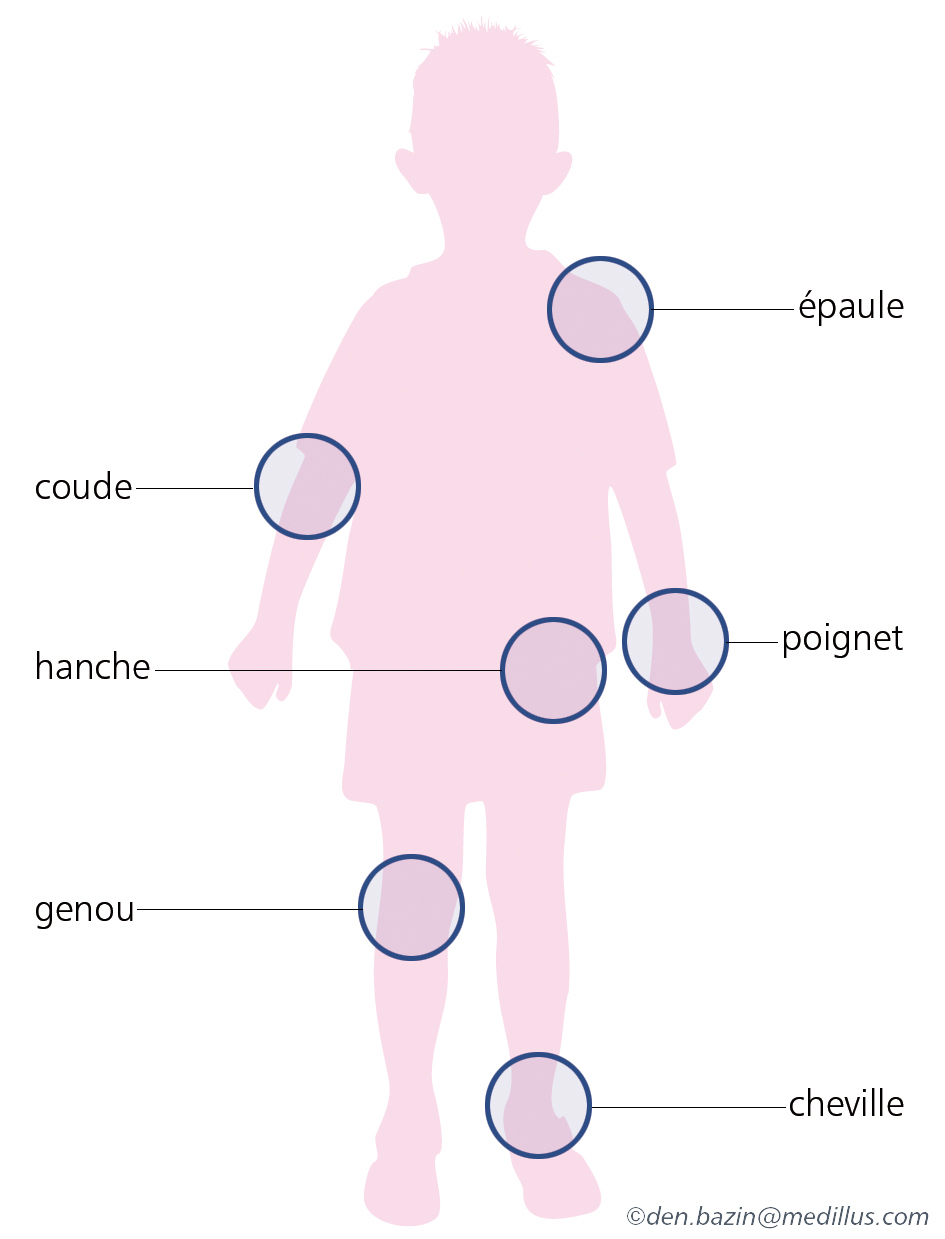
Les localisations dangereuses (cliquer pour agrandir)
- Arthropathie hémophilique : les saignements répétés sur une même articulation entraînent une accumulation de fer contenu dans le sang au sein de la membrane synoviale (mince paroi très vascularisée tapissant l’intérieur de l’articulation) et une augmentation de la pression intra-articulaire. Celles-ci conduisent à l’inflammation de la membrane synoviale et à une destruction du cartilage. L’hyper vascularisation induite dans la membrane synoviale provoque alors une répétition des hémarthroses sur l’articulation cible, ainsi le saignement articulaire devient un cercle vicieux. Plus l’articulation est endommagée, plus les saignements augmentent. Cette arthropathie se traduit par une diminution des mouvements, un handicap fonctionnel et une douleur à la mobilisation, voire au repos.
Les complications articulaires liées aux hémarthroses répétées peuvent conduire à des interventions chirurgicales qui ont pour but de stabiliser ou de remplacer l’articulation abîmée.
- Hémorragies des cavités naturelles :
- Les hématuries : les hématuries sont des événements fréquents, spontanés chez les hémophiles. Le plus souvent, ces hématuries n’ont pas de cause particulière mais il est conseillé de rechercher systématiquement la cause en cas d’hématurie récidivante, d’infection urinaire, de lithiase, d’hypertrophie de la prostate, voire de tumeur.
- Les hémorragies buccales : ces hémorragies sont les plus fréquentes chez l’enfant (hémorragie du frein de la langue ou de la lèvre supérieure, plaie de la muqueuse, morsure de la langue).
- Rectorragies : Les rectorragies résultent de la présence de sang dans les selles, signe généralement d’une hémorragie digestive basse.
- Hématémèses : Ce sont des hémorragies extériorisées par vomissements et résultent d’une hémorragie digestive haute.
- Hémorragies du système nerveux central : elles sont le plus souvent liées à un traumatisme crânien et nécessitent une prise en charge rapide avec injection d’un traitement anti-hémophilique en urgence.
Diagnostic de l’hémophilie+
En première intention, le bilan de dépistage d’une anomalie de la coagulation se fait par des tests dits «globaux » et comporte les analyses suivantes :
- TCA (Temps de Céphaline Activée) explore les facteurs VIII, IX, XI, XII ainsi que les facteurs X, V et II
- TQ (temps de Quick) explore les facteurs VII, X, V et II
- Fibrinogène
- Numération des plaquettes
Le premier signe d’un déficit en facteur VIII ou IX de la coagulation est l’allongement du TCA. A l’inverse le TQ, le fibrinogène et la numération des plaquettes sont normaux.
Devant un TCA allongé, des analyses complémentaires doivent être demandées afin d’éliminer toutes autres hypothèses et pour confirmer la suspicion d’hémophilie.
- Recherche d’anticoagulant
- Dosage spécifique des facteurs XI et XII
- Les dosages des facteurs VIII et IX
Dosage du facteur de Willebrand : le facteur Von Willebrand protége le facteur VIII de la dégradation enzymatique, il est donc important de doser ce dernier lors d’un diagnostic de l’hémophilie.
Le diagnostic prénatal
Dans le cas où la mutation génétique est connue dans la famille, un diagnostic prénatal (avant la naissance) peut être réalisé. Dans un premier temps un diagnostic de sexe sera réalisé par une prise de sang à partir de la 10ème semaine de grossesse ; s’il s’agit d’une fille les investigations s’arrêtent. Dans le cas d’un fœtus de sexe masculin et à la demande des parents, le diagnostic se poursuit par prélèvement de cellules fœtales à la recherche de l’anomalie génétique. Ce prélèvement peut se peut se faire par ponction de villosités choriale sou par amniocentèse. Si le fœtus est diagnostiqué hémophile sévère, une interruption de grossesse peut être proposée.
Dans le cas d’une hémophilie sévère, il est également possible de réaliser un Diagnostic Pré-Implantatoire (DPI) dans le cadre d’une fécondation in vitro. Ce diagnostic est réalisé sur les embryons conçus après fécondation in vitro en prélevant 1 à 2 cellules au 3e jour du développement. Seuls les embryons indemnes sont ensuite implantés dans l’utérus maternel.
Les traitements de l’hémophilie+
Actuellement, il n’existe pas de traitement pour guérir l’hémophilie, mais il existe des traitements efficaces pour prévenir et traiter les accidents hémorragiques. Les traitements à utiliser dépendent d’une part de la sévérité de l’hémophilie et d’autre part du contexte de leur utilisation.
Le traitement des hémophiles sévères et des hémophiles modérés
Le traitement consiste en l’injection par voie intra veineuse du facteur manquant (facteur VIII pour l’hémophile A et facteur IX pour l’hémophilie B). Ces médicaments sont fabriqués à partir de dérivés de sang humain (facteurs plasmatiques) ou par génie génétique (facteur recombinant). Les méthodes de fabrication des médicaments se sont perfectionnées pour garantir la meilleure sécurité permettant l’élimination de virus.
Il est proposé 2 schémas thérapeutiques :
- La prophylaxie, utilisée de préférence pour les hémophiles sévères ainsi que les hémophiles modérés à symptomatologie sévère, consiste à injecter le facteur manquant de manière préventive par 1, 2 ou 3 injection(s) par semaine selon les besoins. Le but est de transformer l’hémophilie sévère en hémophilie modérée sans que le facteur VIII ou IX ne revienne à son niveau de base, et ainsi de limiter les accidents hémorragiques. Le traitement en prophylaxie est le plus couramment utilisé dans les formes sévères de la maladie. Il est généralement débuté chez l’enfant :
- en prévention primaire, avant 2 ans, au moment de l’acquisition de la marche avant ou dès le premier saignement,
- ou en prévention secondaire, après l’âge de 2 ans, suite à la survenue de plus de 2 accidents hémorragiques importants (hématomes ou hémarthrose).
- Le traitement à la demande, utilisé de préférence pour les hémophiles modérés et pour le traitement des saignements de l’hémophilie sévère consiste en l’injection du facteur manquant le plus rapidement possible lors d’un accident hémorragique afin de prévenir au mieux les séquelles.
Une ou plusieurs injections peuvent être nécessaires pour stopper le saignement.
Le traitement des hémophiles mineurs
Le traitement se fait essentiellement lors d’un évènement hémorragique ou avant la réalisation d’un geste chirurgical.
Dans le cas de l’hémophilie A mineure, la desmopressine est utilisée avec de bons résultats (Acétate de desmopressine ou DDAVP). La desmopressine est une forme synthétique de l’hormone naturelle vasopressine qui aide à libérer le facteur VIII de son lieu de stockage dans l’organisme permettant ainsi d’augmenter suffisamment leur taux de facteur VIII et de ne pas avoir à utiliser un facteur de coagulation. Ce médicament peut être administré en intraveineux (Minirin®) ou par pulvérisation nasale (Octim®) lors d’un évènement hémorragique modéré. Une restriction hydrique devra être observée en cas d’utilisation de ce médicament. Dans certaines situations, le traitement par Desmopressine peut s’avérer insuffisant, il convient alors de réaliser une ou plusieurs injections de facteur VIII.
En cas d’une hémophilie B modérée ou mineure, il n’existe pas de traitement dont les propriétés favorisent l’augmentation du taux de facteur IX. La prise en charge thérapeutique d’un évènement hémorragique est donc réalisée par l’injection de facteur IX (plasmatique ou recombinant).
Les facteurs de coagulation commercialisé et en phase de développement sont présentés ici.
Les autres traitements
Les saignements du nez et de la bouche liés ou non à une extraction ou un soin dentaire ainsi que les saignements menstruels peuvent être prévenus et/ou traités à l’aide de deux médicaments qui contribuent à favoriser la stabilité d’un caillot : l’acide tranexamique (Exacyl®) et l’acide aminocaproïque. Ce traitement peut être complété selon le cas par une injection de facteur VIII ou IX.
Le traitement lors des chirurgies en urgence et programmées
En cas d’intervention chirurgicale d’urgence ou programmée, le traitement anti-hémophilique est adapté en fonction du risque hémorragique du geste et de la sévérité de la maladie. Le taux de facteur à atteindre varie en effet en fonction du risque hémorragique de la chirurgie.
Pour les chirurgies à risque hémorragique majeur telles que les neurochirurgies, chirurgies orthopédiques osseuses ou articulaires, chirurgies thoraciques, vasculaires ou cardiaques, certaines chirurgies ORL ophtalmologiques et de stomatologies, le niveau de substitution visera à atteindre un taux de FVIII ou FIX circulant de 80% à 110% pour la première semaine et 50 à 80 % pour les 2èmes et 3èmes semaines suivant la chirurgie.
Pour les chirurgies à risque hémorragique modéré telles que les chirurgies viscérales, urologiques, orthopédiques extravasculaires (synovectomies) d’autres chirurgies ORL ophtalmologiques et de stomatologies, le niveau de substitution visera à atteindre un taux de FVIII ou FIX circulant de 50 à 80 %.
Dans les 2 cas, une injection de FVIII ou FIX est réalisée avant le geste afin d’augmenter le taux de facteur au niveau requis et d’éviter une hémorragie.
« Des conseils de substitution » seront transmis par le médecin traitant l’hémophilie à l’équipe de chirurgie.
La douleur, quant à elle, sera traitée préférentiellement par des antalgiques de classe 1 à 3 selon la sévérité.
Développement des inhibiteurs+
Un inhibiteur est une réponse immunitaire de l’organisme qui produit un anticorps dirigé contre le facteur VIII ou le facteur IX injecté lors de du traitement anti-hémophilique conduisant à la suppression de l’action du facteur de coagulation. Le développement d’inhibiteurs contre un facteur de coagulation est la complication la plus grave du traitement de l’hémophilie. Elle apparaît dans la majorité des cas chez de jeunes enfants dans les 50 premiers jours d’exposition au traitement encore appelés JCPA (journées cumulées de présence de l’antigène). Généralement ceci correspond aux 50 premières injections de traitement. 15 à 30% des hémophiles A développent un inhibiteur contre environ 5% des hémophiles B.
Certains facteurs de risque, tels que les facteurs génétiques, peuvent exercer une influence sur le développement des inhibiteurs.
Parmi les facteurs de risque génétique, le type de mutation touchant le gène du FVIII doit être pris en compte. La situation apparaît différente selon le degré de sévérité de l’hémophilie. L’influence exercée par le type de concentré de F VIII est également citée cependant elle reste un sujet largement discutée.
Diagnostic de l’inhibiteur
La présence d’un inhibiteur est découverte :
- Lorsque le traitement devient moins efficace.
- Lors du suivi biologique comprenant le titrage de l’inhibiteur (en unités Bethesda). Ce titrage est fait en systématique environ toutes les 5 injections jusqu’au 50ème jour d’exposition, puis une fois par trimestre jusqu’au 150ème jour d’exposition.
Traitement de l’inhibiteur
L’inhibiteur peut être neutralisé avec la mise en place d’une induction de tolérance immune (ITI) qui consiste à injecter le traitement anti-hémophilique habituel à de fortes doses de manière répétée et souvent quotidiennement afin de provoquer un essoufflement de la production de l’inhibiteur par l’organisme.
Le traitement des accidents hémorragiques consiste en l’administration de fractions coagulantes activées qui contournent l’action du facteur VIII ou du facteur IX en activant une autre voie dans la cascade de coagulation.
Le choix du traitement se fait selon l’avis du centre de traitement de l’hémophilie en fonction du patient et du titre de son inhibiteur.
Le cas de l’hémophilie acquise
L’hémophilie acquise est une pathologie auto-immune rare avec une prévalence d’environ 1 à 1,5 cas/million d’individu par an). Elle survient généralement après l’âge de 50 ans et touche à la fois les hommes et les femmes. Le système immunitaire se dérègle et entraîne la diminution brutale du facteur de la coagulation VIII détruit par des anticorps anti-facteur VIII. Dans la moitié des cas, la cause de ce dérèglement survient sans raison particulière et dans l’autre moitié des cas, la cause est souvent reliée à une maladie auto-immune, à une tumeur ou à la suite d’une grossesse.
Etat des lieux de la recherche clinique+
La Recherche, très active ces dernières années, donnera lieu à des avancées considérables dans le traitement des hémophiles, une fois que les médicaments à l’essai seront sur le marché. Ces médicaments actuellement en phase de recherche clinique permettront en effet d’améliorer la qualité de vie et le quotidien des patients, parmi lesquels :
- La réduction de la fréquence des injections par la mise à disposition de médicaments à demi-vie prolongée. Ces traitement dont l’efficacité thérapeutique est au moins identique aux molécules conventionnelles permettront d’espacer les injections intraveineuses de façon très significative pour le facteur IX (jusqu’à 1 injection toutes les 2 semaines) et de façon moins marquante pour le facteur VIII (jusqu’à 1 injection par semaine).
- La modification de la voie d’administration du médicament en abordant la voie sous-cutanée par de nouvelles approches :
- Le remplacement du facteur de coagulation manquant par un anticorps monoclonal qui reproduit l’effet normal du facteur VIII activé dans la cascade de la coagulation. Ce médicament destiné aux hémophiles A avec et sans inhibiteur permet d’aboutir à une bonne coagulation en cas de saignement et à une amélioration de la qualité de vie.
- La régulation du système de coagulation par l’inhibiteur physiologique le plus puissant : l’antithrombine.
- La régulation du système de coagulation en inhibant l’activité du TFPI ou INFT (Tissue Factor Pathway Inhibitor ou Inhibiteur de la Voie du Facteur Tissulaire), inhibiteur de la coagulation circulant dans le plasma, par un anticorps monoclonal humanisé.
Pour l’ensemble de ces avancées thérapeutiques, il faudra toutefois attendre la fin des essais cliniques de phase III pour être certain de l’efficacité et de la tolérance de ces produits thérapeutiques.
- La thérapie génique : Plusieurs essais cliniques de thérapie génique sont en cours, utilisant une seule injection intraveineuse d’un adénovirus associé, un vecteur viral recombinant véhiculant le gène du facteur VIII ou du facteur IX. L’injection a permis aux hémophiles B sévères d’obtenir un taux de facteur circulant d’un hémophile jusqu’à 80% et pour les hémophiles A sévères jusqu’à plus de 200%, et ainsi de passer d’un profil d’hémophile sévère à hémophile mineur pour les sujets les plus efficacement traités.