




Le rôle du diagnostic génétique
Dans l’hémophilie
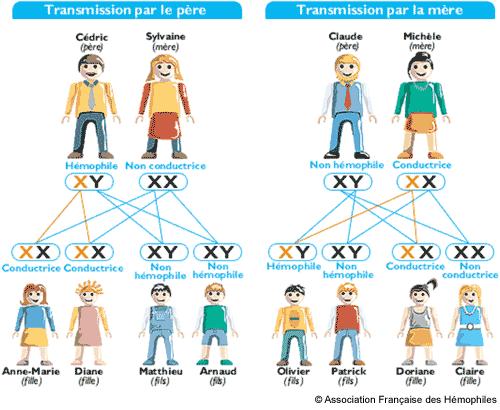
L’hémophilie est une maladie génétique et héréditaire qui se transmet avec le chromosome X.
Les garçons portent un chromosome Y donné par leur père et un chromosome X donné par leur mère, tandis que les filles reçoivent deux chromosomes X, l’un provenant de leur père et l’autre de leur mère. Chez les filles, qui ont deux chromosomes X, l’anomalie du gène situé sur un chromosome X est en général compensée complètement ou partiellement par l’autre chromosome X, sain. A de rares exceptions près, elles ne seront pas malades mais conductrices de l’anomalie qu’elles pourront transmettre à leur descendance. Les garçons ne peuvent pas compenser l’anomalie du gène situé sur le chromosome X, puisqu’il est unique, ils expriment donc la maladie. A noter qu’un tiers des hémophilies portent une mutation de novo, c’est à dire que cette mutation n’est présente ni chez le père ni chez la mère mais qu’elle s’est produite lors de la formation des ovules ou des spermatozoïdes des parents ou plus tard chez le foetus. Cette mutation pourra être transmise à nouveau à la descendance si elle est survenue au niveau des ovules ou des spermatozoïdes des parents.
Pour plus de renseignements : https://www.orpha.net/data/patho/Pub/fr/Hemophilie-FRfrPub646.pdf
L’intérêt du diagnostic moléculaire dans l’hémophilie réside en plusieurs points :
- Le conseil génétique
Il s’adresse à toutes les femmes ayant dans leur famille une personne atteinte d’hémophilie et aux patientes qui présentent un taux de facteur VIII ou IX abaissé sans antécédent familial d’hémophilie connu. Dans un tiers des cas, les conductrices ont un bilan d’hémostase normal. Seule l’étude génétique permet la détermination avec certitude du statut chez toutes les femmes à risque. Ceci nécessite au préalable l’identification de la mutation responsable de la maladie chez un apparenté atteint.
La détermination du statut de conductrice d’hémophilie peut mener à une démarche de diagnostic prénatal dans l’éventualité d’un fœtus de sexe masculin. Pour cette raison, l’étude moléculaire doit être réalisée le plus rapidement possible avant une éventuelle grossesse.
- Le diagnostic prénatal (diagnostic fait avant la naissance)
Il commence par le diagnostic prénatal non invasif du sexe fœtal. Ceci est réalisé par l’étude précoce du fœtus sur une simple prise de sang de la mère en général vers la Xème semaine de grossesse. Cette technique décrite en 2002 est recommandée par la Haute Autorité de Santé car elle ne fait courir aucun risque à la grossesse en cours.
Dans le cas d’un fœtus de sexe masculin et sur la demande des parents, il est alors nécessaire d’obtenir des cellules fœtales pour rechercher l’anomalie génétique et savoir si le fœtus est atteint ou non. Le prélèvement fœtal est obtenu par ponction de villosités choriales ou de liquide amniotique selon le terme de la grossesse.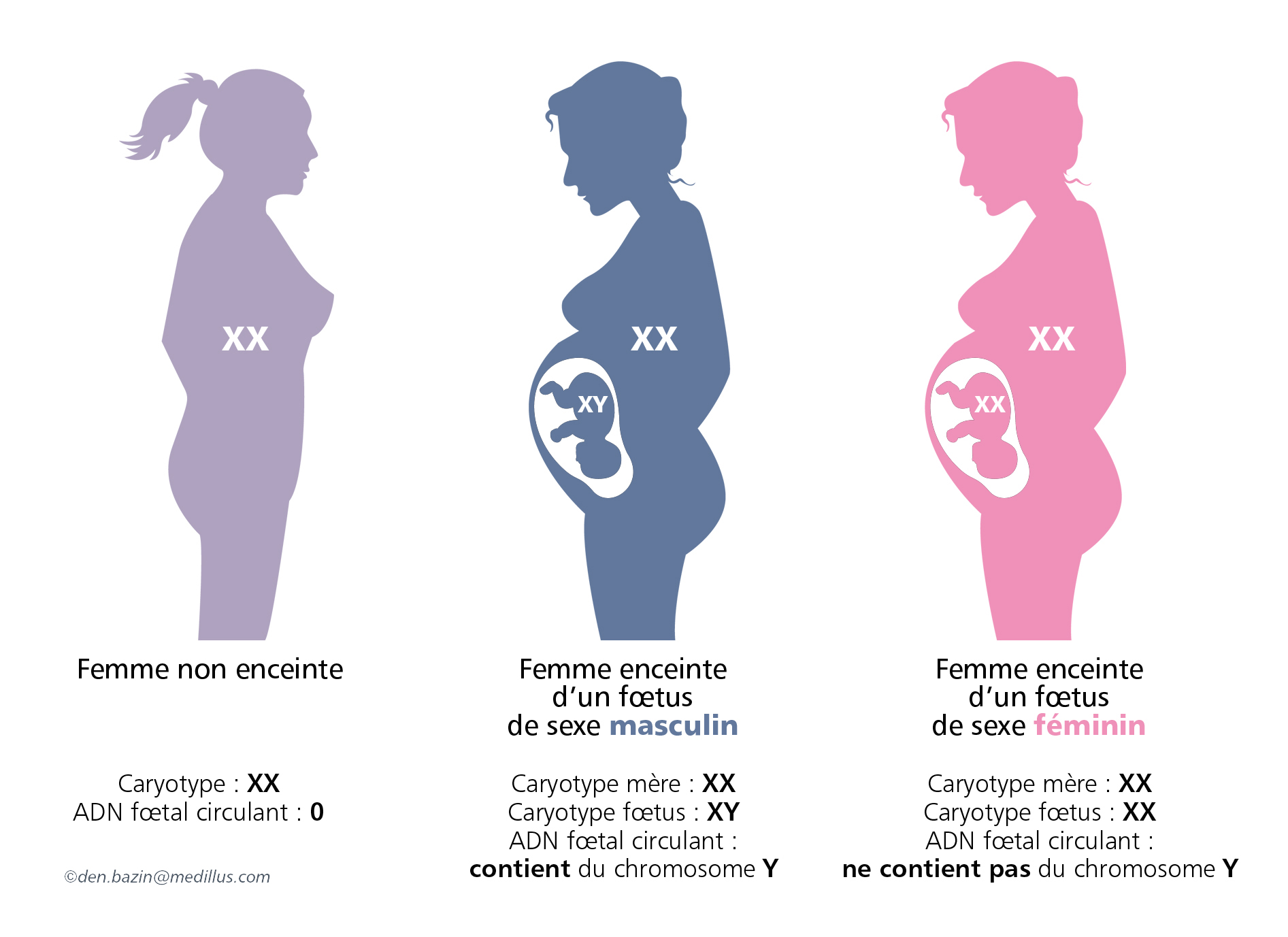
- Le diagnostic préimplantatoire
Le diagnostic préimplantatoire (DPI) offre aux couples, ayant un risque élevé de transmettre une maladie héréditaire sévère, une alternative au diagnostic prénatal (DPN) et l’éventuelle interruption médicale de grossesse qui s’ensuit parfois. Il est effectué à partir d’une ou deux cellules prélevées sur un embryon âgé de trois jours, issu d’une fécondation in vitro (FIV). Il consiste à rechercher l’anomalie génétique responsable de la maladie dans la famille, afin de ne transférer chez la patiente que les embryons sains.
Pour plus de renseignements : https://www.inserm.fr/thematiques/biologie-cellulaire-developpement-et-evolution/dossiers-d-information/assistance-medicale-a-la-procreation
- Les bases de données de mutations
Ces bases sont enrichies régulièrement par la description de la mutation identifiée chez les patients et de leurs signes cliniques tels la sévérité et la survenue d’inhibiteur (facteur de résistance au traitement). Ces données sont une aide précieuse pour les patientes sans antécédents familiaux d’hémophilie, ayant un taux abaissé de facteur, faisant suspecter une conduction d’hémophilie. L’étude génétique permet parfois, de détecter une mutation. Si elle est décrite dans les bases de données, il est alors fort probable que l’hémophilie transmise par cette femme sera du même degré de sévérité. Mais il est par contre difficile d’établir un diagnostic dans le cas d’une mutation jamais décrite, d’où l’importance d’étudier tous les patients hémophiles quelques soit leur sévérité. Ces bases permettent également d’alerter sur le risque de survenue d’un inhibiteur chez les patients nouvellement diagnostiqués.
- La relation mutation et survenue d’anticorps anti facteur VIII ou IX (ou inhibiteur) et prise en charge du patient hémophile
Le développement d’inhibiteur est plus fréquent dans l’hémophilie A (20% à 30%) que dans l’hémophilie B (5%). Il survient le plus souvent dans les formes sévères mais est aussi décrit dans les formes modérées et mineures avec parfois un taux d’anticorps très élevé comparable à celui observé dans les hémophilies A sévères. L’anomalie génétique responsable de la maladie est un facteur de risque de développement d’inhibiteur et il existe une corrélation entre l’anomalie génétique et le risque de développer un inhibiteur avec une incidence/ prévalence différente en fonction du type de mutation. Même si le développement d’un inhibiteur est un phénomène multifactoriel, la connaissance d’un génotype « à risque » peut inciter à une surveillance accrue ou à une attitude thérapeutique différente.
Dans les autres déficits constitutionnels en facteur de la coagulation
Il s’agit de déficits rares voire très rares dont le diagnostic se fait également par le dosage des facteurs concernés. La maladie touche aussi bien les hommes que les femmes avec une prise en charge particulière de la grossesse et de l’accouchement. L’analyse moléculaire est possible dans tous les déficits mais n’est pas indispensable au diagnostic.
Le diagnostic moléculaire permet de proposer un diagnostic prénatal dans les familles à risque de transmission pour les déficits sévères. Il nécessite l’identification de la mutation causale chez les deux membres du couple. Tous les déficits en facteur de la coagulation ne sont pas associés à des formes cliniques sévères, ce sont essentiellement les déficits en FX, FVII, FXIII, les déficits combinés en Facteurs vitamine K dépendants et l’afibrinogénémie. Le déficit en FII ne serait pas viable dans sa forme la plus sévère. Le déficit en FXI ne s’accompagne pas de tableaux cliniques à type d’hémorragies intracérébrales justifiant un diagnostic prénatal. Ces tableaux sont aussi extrêmement rares en cas de déficit en FV. Ainsi, le conseil génétique dépend du retentissement clinique de l’affection dans une famille donnée. Seule l’existence d’un premier enfant atteint de manifestations très sévères peut conduire à proposer un diagnostic anténatal lors d’une grossesse ultérieure. En effet, les formes éligibles au diagnostic prénatal, pour l’ensemble des déficits rares, restent exceptionnelles. La plupart des déficits rares en facteurs de la coagulation même ceux en FVII, FX ou FXIII sont dans l’immense majorité des cas compatibles avec une vie normale.
Le diagnostic génétique est également utile dans des cas bien particuliers. Dans les déficits sévères en FXI, par exemple, il permet d’évaluer le risque de développer des inhibiteurs et ainsi d’orienter la conduite à tenir. Dans les anomalies du fibrinogène, certaines mutations sont associées à un risque accru de thromboses et sont importantes à dépister.
Enfin il permet de clarifier certaines situations complexes et de poser le diagnostic, notamment en cas de déficits combinés (FVII, FX, FV et FVIII etc…).
L’identification des défauts moléculaires et leur caractérisation fonctionnelle ont permis de progresser de façon notable dans la relation structure-fonction de la molécule.
Comme pour l’hémophilie, il existe des bases de données des mutations dédiées à chaque type de déficit. Elles sont des outils indispensables au biologiste moléculaire comme au clinicien pour l’interprétation des mutations. Cependant, il existe une difficulté supplémentaire car les déficits rares en facteur de la coagulation sont de transmission autosomique récessive, c’est-à-dire qu’il faut que les deux chromosomes soient touchés pour que la maladie s’exprime. Ainsi, de nombreux patients présentent deux mutations différentes, chacune ayant son propre mécanisme et ses propres conséquences rendant l’interprétation de la combinaison des deux (génotype) complexe.
Dans la maladie de Willebrand (VWD)
L’analyse génétique permet d’aider au diagnostic et surtout d’anticiper la prise en charge des patients pour des formes de la pathologie particulières (types 2A, 2B, 2N et 1). Elle doit être faite précocement pour identifier les patients avec VWD sévère (type 3) qui ont un risque important de développer un anticorps anti-VWF pour ajuster leur prise en charge clinique. Pour les patients de groupe O la mise en évidence de mutations d’interprétation claire du gène VWF permet d’affirmer le diagnostic. Un diagnostic prénatal n’est proposé que dans les formes sévères de maladie de Willebrand de type 3. Ainsi l’identification de la mutation peut éclairer les choix de traitement et améliorer la compréhension des mécanismes sous-jacents des différents sous-types de maladie de Willebrand. Il est cependant évident que les tests génétiques ont une utilité clinique limitée dans les cas où le phénotype révèle clairement le type de maladie de Willebrand.