




Les essais thérapeutiques
Qu'est-ce qu'un essai clinique?+
D’après l’Agence Nationale de Santé du Médicament et des produits de santé (ANSM), un essai clinique est une recherche biomédicale organisée et pratiquée sur l’Homme en vue du développement des connaissances biologiques ou médicales.
Lorsque la recherche a pour objectif de tester un nouveau médicament ou de développer un médicament existant (pour une nouvelle indication, une nouvelle voie d’administration…), le terme habituellement employé est celui d’essai thérapeutique.
Les essais cliniques portant sur les médicaments ont pour objectif, selon le cas, d’établir ou de vérifier certaines données pharmacocinétiques (modalités de l’absorption, de la distribution, du métabolisme et de l’excrétion du médicament), pharmacodynamiques (mécanisme d’action du médicament notamment) et thérapeutiques (efficacité et tolérance) d’un nouveau médicament ou d’une nouvelle façon d’utiliser un traitement connu.
Un essai thérapeutique est soumis à des exigences réglementaires très strictes. Pour débuter les inclusions dans un centre, le promoteur, personne physique ou morale qui prend l’initiative de l’essai clinique, doit impérativement obtenir :
- Avis favorable du CPP (Comité de Protection des Personnes). Le CPP a pour rôle des’assurer que tout projet de recherche biomédicale sur l’être humain mené en France respecte diverses mesures (médicales, éthiques et juridiques) visant à assurer la protection des personnes qui participeront à cette recherche
- Avis favorable/autorisation de la CNIL (Commission Nationale Informatique et Libertés) : cet organisme veille à ce que les traitements de données à caractère personnel soient mis en œuvre conformément aux dispositions de la loi n°78-17 du 6 janvier 1978 relative à l’informatique, aux fichiers et aux libertés
- Autorisation de l’ANSM : l’ANSM s’assure que toutes les conditions de sécurité soient réunies pour la conduite de l’essai sur le territoire français
Une assurance : Depuis une loi de 1988, les promoteurs réalisant des essais cliniques en France ont l’obligation de souscrire une assurance responsabilité civile ad hoc.
Le chemin du médicament en France+
Le développement d’un nouveau médicament ou l’amélioration d’un médicament déjà existant, de la molécule à sa commercialisation, est un processus long nécessitant dix à quinze ans de recherche en moyenne.
Ce développement nécessite un nombre d’étapes (ou phases) défini et strictement réglementé.
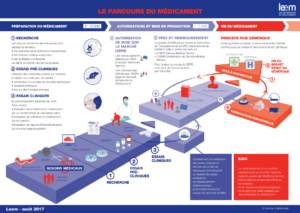
Source : www.leem.org
1. La recherche cognitive ou « Drug Discovery »
Cette première étape a pour objectif de découvrir des candidats médicaments, qui seront ensuite testés au cours du développement préclinique, puis clinique. C’est la première des grandes phases de la recherche et développement.
Le principe de la recherche cognitive consiste à mettre au point une série de composés actifs d’origine chimique ou biologique testés dans des modèles expérimentaux in vitro ou ex vivo sur un mécanisme pathologique. Au cours de ce criblage, les composés actifs interagissant sur la cible sont sélectionnés, puis ceux qui agissent plus spécifiquement sur le cible sont optimisés de façon à les rendre administrables in vivo.
Ainsi, plus de 100 000 molécules vont être criblées, pour aboutir à 200-250 molécules d’intérêt qui vont poursuivre le processus de développement (préclinique puis clinique).
2. La phase préclinique
Le développement préclinique consiste à évaluer, in vivo sur des systèmes vivants non humains, l’activité d’un candidat médicament issu des phases de la recherche cognitive.
Le développement préclinique fait en particulier appel à l’expérimentation animale, qui est une étape indispensable à la connaissance d’un futur médicament avant de l’administrer à l’homme. En effet il n’est pas envisageable d’administrer, dans un premier temps, un nouveau composé à l’homme sain ou malade compte tenu des risques non connus susceptibles d’apparaître. L’expérimentation animale est donc utilisée de manière rationnelle et, dans tous les cas, selon des bonnes pratiques qui garantissent un traitement éthique de l’animal de laboratoire.
Au cours du développement préclinique, un grand nombre d’études est effectué afin de qualifier le candidat médicament sur le plan de la pharmacologie, de la pharmacocinétique (modalités de l’absorption, de la distribution, du métabolisme et de l’excrétion du médicament) et de la toxicologie (détermination des seuils de toxicité/létalité). Ces études sont constitutives d’une partie du dossier de demande d’Autorisation de Mise sur le Marché (AMM) du futur médicament ; elles répondent à des normes internationales de qualité scientifique et sont étroitement évaluées par les autorités de santé au moment de délivrer l’AMM.
Parmi les 200 à 250 molécules initialement testées dans ces phases précliniques, seules environ dix arriveront aux phases cliniques.
3. Les phases cliniques
La phase I
A ce stade, les essais sont menés principalement sur un nombre limité de sujets sains, sous strict contrôle médical. Ces volontaires peuvent être indemnisés. La molécule est testée sur une courte période.
L’objectif est d’évaluer :
- La sécurité d’emploi du produit
- Son devenir dans l’organisme
- Son seuil de tolérance
- Les effets indésirables
Dans certains domaines comme l’oncologie, ou plus près de nous, les maladies hémorragiques, la phase I est effectuée sur des patients souffrant de la pathologie ciblée.
La phase II
Les essais sont réalisés sur un nombre limité de patients volontaires. Leur objectif est de tester l’efficacité du produit, de déterminer la dose optimale (posologie) et d’observer les effets secondaires éventuels.
Ce sont ces essais qui conditionnent la mise en place ou non d’études à plus grande échelle : la phase III.
La phase III
Menés sur de larges populations de patients, les essais de phase III permettent de comparer l’efficacité thérapeutique de la molécule au traitement de référence (lorsque celui-ci existe) ou bien à un placebo (lorsqu’aucune thérapie n’existe). Ces essais sont très souvent multicentriques , menés dans de nombreux centres d’études, souvent à l’international.
Des études « ancillaires » (qui découlent de l’étude principale) peuvent être développées pour examiner des populations de patients particulières (la chirurgie, la pédiatrie,…)
Durant ces phases, se déroulent également des essais relatifs au développement industriel et au mode d’administration et de conditionnement (gélules, comprimés, solution injectable…).
La durée de ces études peut parfois atteindre plusieurs années.
Les dossiers d’AMM et ANSM
Tous les résultats recueillis lors des recherches cognitives, des essais précliniques et cliniques vont constituer le dossier d’Autorisation de Mise sur le Marché (AMM), déposé par les laboratoires pharmaceutiques.
Pour être commercialisé, tout médicament fabriqué industriellement doit faire l’objet d’une AMM.
L’AMM peut être délivrée au niveau européen par la Commission européenne, après avis favorable du Comité des Médicaments à Usage Humain (CHMP) de l’Agence Européenne du Médicament (EMA). Au niveau national, c’est l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) qui examine les demandes de mise sur le marché. Le nouveau médicament doit présenter un rapport bénéfices/risques au moins équivalent à celui des produits déjà commercialisés.
L’accès au remboursement se fait dans un second temps, à l’initiative des laboratoires pharmaceutiques qui soumettent leur demande à la Commission de la Transparence de la HAS (Haute Autorité de Santé). L’avis de cette commission est ensuite transmis au Comité Economique des Produits de Santé (CEPS) qui détermine le prix du médicament et à l’Union nationale des caisses d’assurance maladie (Uncam) qui en fixe le taux de remboursement.
La décision finale d’inscription relève de la compétence du ministre de la santé et est publiée au Journal officiel.
Une fois commercialisé, le médicament reste sous surveillance. Ainsi, le rapport bénéfices/risques du produit est évalué en permanence pour prendre la mesure des effets indésirables connus ou nouvellement identifiés.
En cas de risque pour la santé, un médicament peut se voir appliquer une décision de police sanitaire prenant la forme d’une restriction ou d’une modification des indications. Le médicament peut également faire l’objet d’un retrait du marché.
La phase VI
Les essais ne s’achèvent pas avec l’autorisation de mise sur le marché, mais se ils poursuivent tout au long de la commercialisation du médicament. Des essais dits de Phase IV, sont réalisés dans des conditions proches de la prise en charge habituelle. Ces essais ont pour objectifs de repérer d’éventuels effets indésirables rares non détectés durant les phases précédentes (pharmacovigilance) et de préciser les conditions d’utilisation pour certains groupes de patients à risques. Cette phase permet d’analyser les interactions médicamenteuses et favorise la mise au point de nouvelles formes galéniques (gélule, cachet, solution injectable, crème, gel…) ainsi que des extensions d’indications thérapeutiques.
4. Cas particulier : l’Autorisation Temporaire d’Utilisation (ATU)
En France, l’utilisation exceptionnelle de spécialités pharmaceutiques ne bénéficiant pas encore d’une AMM, ou utilisées dans une indication non prévue par l’AMM et ne faisant plus l’objet d’un essai clinique est conditionnée à l’obtention préalable d’une Autorisation Temporaire d’Utilisation (ATU).
Les ATU sont délivrées par l’ANSM dans les conditions suivantes :
- Les spécialités sont destinées à traiter, prévenir ou diagnostiquer des maladies graves ou rares.
- Il n’existe pas de traitement approprié.
- Leur efficacité et leur sécurité d’emploi sont présumées en l’état des connaissances scientifiques.
En pratique, il existe deux types d’ATU :
- Les ATU de cohorte qui s’adressent à un groupe ou sous-groupe de patients traités et surveillés suivant des critères définis dans un protocole d’utilisation thérapeutique et de recueil d’informations.
- Les ATU nominatives qui s’adresse à un seul patient nommément désigné et ne pouvant participer à une recherche biomédicale.
Financement+
Les essais thérapeutiques ayant pour but de développer un nouveau médicament, ou modifier un médicament existant en vue d’une commercialisation, sont pour la plupart financés par l’industrie pharmaceutique. Dans les centres français spécialisés sur les maladies hémorragiques constitutionnelles, 92,3% des essais thérapeutiques ont été financés par des industries pharmaceutiques en 2016.
Dans une moindre proportion, des essais thérapeutiques peuvent être financés par des institutions publiques ou privées. Dans ces cas-là, le financement est souvent pluriel : financement par l’industrie pharmaceutique + financement institutionnel (Programmes Hospitaliers de Recherche Clinique, Bourses, etc.). En 2016, 30,8% des centres d’hémostase ont bénéficié de financement publiques et 7,7% d’institutions privées.
Quel que soit le mode de financement, ni le volontaire/patient participant à un essai, ni la Sécurité Sociale ne doit avoir à assumer une charge financière en lien avec les activités de l’essai thérapeutique.
Les consultations, les examens d’imagerie, les bilans biologiques, le médicament à l’essai, les frais de transport ainsi que tout le matériel nécessaire à la prise du médicament comme les injections doivent être pris en charge par le laboratoire ou l’institution promoteur de l’essai.
Participer à un essai thérapeutique+
La participation volontaire à un essai clinique est utile à tous : pour soi-même, pour la communauté scientifique et médicale, pour les autres patients, afin de permettre à tous l’accès à des thérapies à chaque fois plus innovantes.
1. A qui s’adresser ?
Les essais thérapeutiques sont en général à l’initiative de l’industrie pharmaceutique. Celle-ci dispose des moyens et des équipes pour la recherche préclinique, ainsi que des moyens de production et de distribution de ses spécialités pharmaceutiques.
Par contre, elle n’a pas un accès direct aux patients. Pour réaliser les essais thérapeutiques, l’industrie pharmaceutique doit donc se rapprocher des médecins. Les essais sont ainsi proposés aux médecins qui choisissent ou non de participer aux différentes études. Si le médecin accepte, alors un processus administratif et règlementaire très strict se met en place pour lui permettre la conduite de l’essai dans l’établissement où il exerce. . Pour débuter les inclusions dans un centre, le promoteur, personne physique ou morale qui prend l’initiative de l’essai clinique, doit impérativement obtenir :
- Avis favorable du CPP (Comité de Protection des Personnes) : le CPP a pour rôle de s’assurer que tout projet de recherche biomédicalesur l’être humain mené en France respecte diverses mesures (médicales, éthiques et juridiques) visant à assurer la protection des personnes qui participeront à cette recherche
- Avis favorable/autorisation de la CNIL (Commission Nationale Informatique et Libertés) : Cet organisme veille à ce que les traitements de données à caractère personnel soient mis en œuvre conformément aux dispositions de la loi n°78-17 du 6 janvier 1978 relative à l’informatique, aux fichiers et aux libertés
- Autorisation de l’ANSM (Agence Nationale de Santé du Médicaments et des produits de santé) : l’ANSM s’assure que toutes les conditions de sécurité soient réunies pour la conduite de l’essai sur le territoire français
- Une assurance Depuis une loi de 1988, les promoteurs réalisant des essais cliniques en France ont l’obligation de souscrire une assurance responsabilité civile ad hoc.
Une fois que le centre est ouvert aux inclusions, le médecin (appelé Investigateur) est en capacité de pouvoir proposer l’essai à ses patients. Il n’est donc pas rare qu’un essai thérapeutique puisse être proposé dans un établissement hospitalier sans l’être dans un autre.
Un patient peut également avoir été informé sur la conduite d’un essai thérapeutique (association de patients, site internet dédié, médias…) et peut aborder ce sujet avec son médecin. Cependant, il lui sera difficile de participer à l’essai clinique si son centre n’est pas ouvert en tant que centre investigateur.
Dans tous les cas, le bon réflexe à avoir est d’aborder le sujet avec son médecin référent qui sera en mesure de se renseigner sur l’essai, sur les modalités de participation, sur les bénéfices/risques, etc.
2. Quelles sont les garanties de protection ?
Les essais thérapeutiques sont menés par des équipes multidisciplinaires compétentes dans la pathologie concernée et/ou dans la conduite d’essais thérapeutiques, dans le service directement de l’investigateur ou dans un service spécialisé (Centre d’Investigation Clinique, Unité de Recherche Clinique…).
Pour rappel : Tout essai thérapeutique, pour pouvoir être conduit, a nécessairement reçu l’approbation d’un comité d’éthique (Comité de Protection des Personnes), l’autorisation de la CNIL, et l’autorisation de l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM). De plus, l’essai est couvert par une Assurance responsabilité civile ad hoc
3. Tout le monde peut-il participer à un essai thérapeutique ?
Non. Les essais thérapeutiques nécessitent un « calibrage » de la population étudiée. Des critères stricts doivent être respectés (critères d’âge, de sexe, de conditions médicales…). Ils différent à chaque étude.
4. Comment se déroule un essai thérapeutique ?
Le médecin « Investigateur » propose l’essai thérapeutique en informant le patient sur l’objet de l’étude, ses objectifs, les éventuels bénéfices attendus et les contraintes imposées par le protocole.
Une notice d’information de l’étude est donnée au patient lui permettant de prendre connaissance de l’étude posément. Un temps de réflexion adéquat doit être laissé au patient pour qu’il puisse réfléchir sereinement à sa participation, qu’il ait l’occasion de se renseigner et d’appréhender les bénéfices attendus mais aussi les contraintes et les éventuels effets indésirables.
Une fois l’essai accepté, les procédures liées au protocole de l’essai ne démarrent qu’après l’obtention orale, écrite et libre du consentement du patient. Un manquement ou une déviation à ce processus expose l’investigateur à de sévères sanctions.
Afin de respecter les conditions de sécurité des patients et d’obtenir des données fiables répondant aux objectifs, les essais thérapeutiques peuvent être contraignants et nécessitent donc une pleine acceptation de ces contraintes à la fois par le patient et par l’équipe médicale :
- Pour le patient, ces contraintes peuvent être liées à un nombre important de visites à l’hôpital, de prélèvements sanguins, un contrôle strict des unités thérapeutiques et un respect total des conditions d’administration et/ou de stockage du produit à l’essai, du remplissage des questionnaires, d’examens complémentaires(imagerie…), etc.
- Pour l’investigateur, en plus des contraintes liées à la fréquence et à l’augmentation de la durée des visites, des contraintes administratives s’ajoutent (processus de recueil de données crédibles et vérifiables, vérification de celles-ci par des tiers, procédures de recueil et transmissions des évènements indésirables graves et non graves, etc.).
5. Le patient peut-il refuser de participer à un essai clinique ?
Oui. Le patient (et /ou les parents pour les enfants mineurs) est (sont) libre(s) de refuser de participer à un essai. La prise en charge du patient auprès de l’équipe médicale n’est absolument pas modifiée en conséquent.
6. Le patient peut-il arrêter de participer à un essai en cours ?
Oui. A tout moment, un patient (ou les parents) peut mettre un terme à sa participation à un essai sans être pour autant obligé de motiver sa décision et sans que sa prise en charge par son médecin en soit modifiée.
De même, un investigateur peut décider de retirer un patient d’un essai, après l’avoir informé, s’il juge que l’intérêt ou la sécurité du patient est mis en cause par celui-ci, ou si le patient ne respecte pas le protocole., . Des procédures de sortie d’essai sont prévues par le protocole, et le suivi médical habituel reprend comme précédemment. L’étude peut également être interrompue si un risque inattendu d’événement indésirable grave survient. Dans ce cas, le patient est informé de l’arrêt prématuré de l’étude et des modalités de fin anticipée pouvant inclure une ou plusieurs visites de contrôle.
7. A la fin de l’étude, le patient peut-il continuer à bénéficier du traitement à l’essai ?
Dans la majorité des cas, les laboratoires pharmaceutiques proposent une étude « d’extension » à la suite d’un essai thérapeutique. Cela permet de pouvoir continuer de proposer le nouveau traitement jusqu’à l’obtention de l’Autorisation de Mise sur le Marché (AMM).
8. Un essai clinique coûte-t-il au patient ? A la Sécurité Sociale ?
Les patients participant à un essai thérapeutique ne doivent subir aucune charge financière en lien direct avec la recherche.
De même, la Caisse Primaire d’Assurance Maladie (CPAM), ne supporte aucune charge financière ni sur la prise en charge des actes liés à la recherche (hospitalisation, temps médical, infirmier,…), ni sur le traitement à l’essai.
Le laboratoire pharmaceutique supporte l’ensemble de ces charges et selon les contraintes liées à l’étude, il peut même être amené à verser une compensation financière au patient.